Authors
Adnan Erol
Abstract
According to the current view, tumor necrosis factor (TNF) receptor 2 (TNFR2) provides neuroprotection and regeneration by activating both canonical and noncanonical NF-κB pathways when activated by paracrine membrane-bound TNF (mTNF). A careful analysis of the literature reveals, however, that mTNF-mediated TNFR2 stimulation only stimulates canonical NF-B signaling, which in turn inhibits the NF-κB-inducing kinase (NIK)-mediated noncanonical pathway. Moreover, microvesicles, which contain transmembrane TNF, secreted by activated microglia in response to danger signals also interact with TNFR2. Therefore, only cytoplasmic TNFR2 can initiate NIK activation following ligand-TNFR2 complex endocytosis. Activated NIK phosphorylates Drp1, promoting mitochondrial fission and inducing oxidative stress. Following that, oxidative stress and/or direct NIK activate c-Abl, a non-receptor tyrosine kinase that can initiate almost any neurodegenerative process. In conclusion, TNFR2 appears to be responsible for neurodegeneration as well while maintaining neuron viability, depending on the ligand.
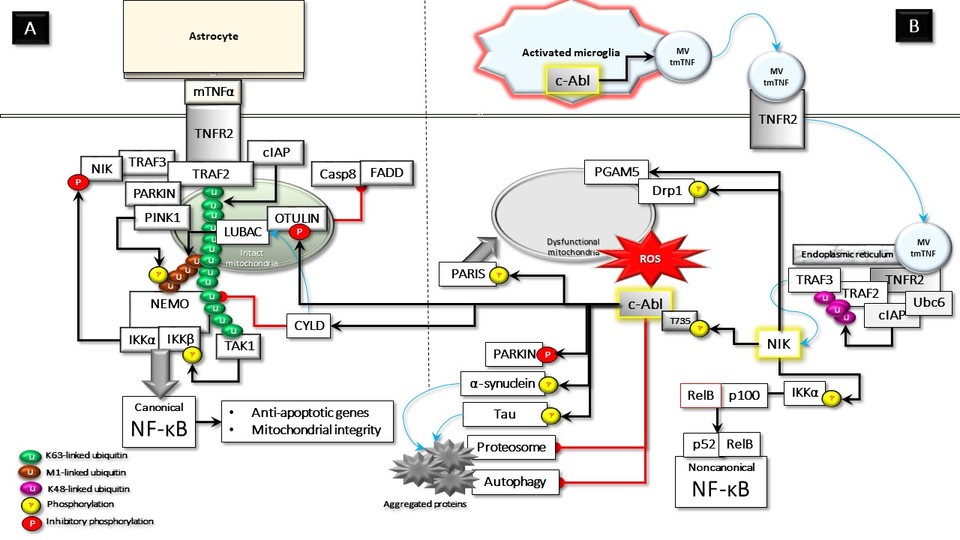 Image 1
Image 1
Main content
Introduction
When activated, TNFR2 signaling is considered to activate both canonical and non-canonical NF-κB signaling pathways, which support cellular viability. Therefore, therapeutic support for activating the TNFR2 pathway is being proposed as one of the leading options for controlling chronic inflammatory degenerative disorders [1]. However, this viewpoint may have some critical flaws that should be carefully examined in the context of neurodegenerative diseases. Although TNFR2 is primarily expressed in immune cells, its expression in both neuronal and glial cells has been validated [1–3]. TNFR2 binds to different ligands, resulting in diverse outcomes at the membrane level and the cytoplasm [1, 4]. While soluble TNF (sTNF) can bind with high affinity, it does not activate intact TNFR2 signaling. Therefore, the natural ligand of TNFR2 is membrane-bound TNF (mTNF) [5]. In resting cells, TRAF2 and TRAF3 interact with cIAPs, probably at the perinuclear endoplasmic reticulum (ER) area. This complex is associated with the E2 protein Ubc6, which is required for the degradative E3 ligase activity of cIAPs [2]; subsequently, it enables the cIAP-mediated ubiquitination and degradation of TRAF3-associated NIK [3]. In response to mTNF paracrine stimulation by glial cells, astrocytes, and, in particular, neuronal TNFR2 oligomerizes in the membrane. Thus, activated TNFR2 recruits the adapter molecule TRAF2 in a complex with TRAF3, cIAPs, and TRAF3-associated NIK [4]. TRAF2 also interacts with Parkin [5] and mitochondria [6]. Parkin, in turn, interacts with both NEMO and LUBAC, which is necessary for the linear ubiquitination of NEMO [7]. Furthermore, NF-κB and its upstream regulatory proteins, including IKKα and IKKβ, were also previously identified in mitochondria [8]. Activated TNFR2 interacts with mitochondria-associated TRAF2. This interaction facilitates the lysine 63 (K63)-linked ubiquitination of TRAF2 by cIAPs [9]. In addition, Parkin-recruited LUBAC, which is associated with OTULIN, decorates the outer membrane of mitochondria with methionine (M)1-linked ubiquitin chains. This promotes the assembly of an NF-κB signaling platform at the mitochondria. Surprisingly, HOIP, the catalytic component of LUBAC, interacts with PINK1 and stabilizes it through the modification of M1-linked ubiquitin chains. The activated PINK1 on mitochondria, in turn, phosphorylates linear ubiquitin chains, preventing their hydrolysis by the deubiquitinase OTULIN without dissipating the mitochondrial membrane potential [10]. Together, K63-linked and M1-linked ubiquitin chains create a docking platform for NEMO and TAK1 [9]. Thus, NEMO and TAK1 activate IKKα and IKKβ for the induction of the canonical NF-κB signaling pathway [1,9]. Subsequently, TNFR2-mediated NF-κB activation can protect neurons by modulating the p300/Stat3/RelA/OPA1 signaling cascade. This can significantly improve mitochondrial integrity and adaptive responses and support neurons’ ability to withstand stress [11]. NFKB activity also induces transcriptional up-regulation of anti-apoptotic genes, which support neural viability. Activated IKKα alone has previously been shown to directly phosphorylate NIK, causing NIK destabilization and thus inhibiting the hyperactivity of the noncanonical NF-kB pathway [12]. However, a later study reported that activating the heterotrimeric IKK complex (NEMO, IKKα, and IKKβ), but not IKKα alone, inhibits the activation of noncanonical NF-B signaling by phosphorylating and inhibits NIK activity [13]. In other words, the canonical NF-kB signaling pathway must be activated to control NIK activity and the associated noncanonical NF-kB pathway. TNF has been identified as a neuroprotective cytokine that prevents neuronal damage under various stress conditions. Furthermore, neuronal survival requires constitutive NF-κB activation via para- or autocrine loops [14,15]. A recent study has excellently shown that after TNF stimulation, intact mitochondria can provide a platform for the activation of NF-κB transcription factors and then transport them to the nucleus [10]. This mode of transport may be particularly important for polarized cells such as neurons. NF-κB regulates energy metabolism networks by balancing glycolysis utilization and mitochondrial respiration [16]. Under normal metabolic conditions, the "resting" brain consumes glucose and metabolizes it almost entirely to CO2 via mitochondrial oxidative phosphorylation [17]. Taken together, TNFR2 signaling induced by paracrine mTNF and the resulting NF-B activation protects neuronal viability and regulates synaptic plasticity [1] (Figure A). An unconventional third mode of TNF signaling is proposed in addition to conventional sTNF and mTNF signaling. In response to danger signals, the transmembrane TNF (tmTNF) is packaged into potent, pro-inflammatory microvesicles (MVs), which protect the enclosed TNF, allowing effective signaling to target cells [18]. Extracellular MVs carrying tmTNF are released by activated microglia interact preferentially with TNFR2 in neurons [18–20] and may form a ligand-receptor complex that undergoes endocytosis [20,21]. TRAF2, TRAF3, and E3 ubiquitin ligase cIAP1/2 are all associated with the cytoplasm, and TRAF3 functions as a NIK binding adapter. The unconventional ligation of TNFR2 translocates this complex to the perinuclear ER compartment where Ubc6 resides. Thus, cIAP and Ubc6 work together to degrade TRAF2 and TRAF3 by catalyzing K48-linked ubiquitination [2,22,23]. TNFR2-induced TRAF2/TRAF3 degradation may release NIK and the accumulated NIK promotes the activation of the noncanonical NF-κB signaling [23]. Altogether, only danger signals-induced unconventional TNF stimulation, but not mTNF may enable TNFR2-mediated NIK accumulation. Consequently, the widely accepted view that TNFR2 stimulation causes simultaneous canonical and non-canonical NF-κB activation may need to be reconsidered. Once NIK is stabilized, it phosphorylates IKKα and results in phosphorylation-dependent ubiquitination of NF-κB precursor protein p100, liberating the p52 subunit, which forms a heterodimer complex with RelB for the activation of noncanonical NF-κB pathway [24]. The cellular outputs of the noncanonical NF-κB pathway are ultimately determined by p52:RelB dimer-mediated gene processing. Hyperactivity of this pathway plays an important role in the etiopathogenesis of some proliferative and autoimmune diseases [25]. Remarkably, RelB expression levels were found to be very low in neurons [26]. Moreover, It has even been claimed that increased NIK expression in neurons, surprisingly, inhibits rather than activates NF-κB. Accordingly, the same study states that glia and other non-neuronal cell types are responsible for almost all of the NF-κB activity observed in the brain [27]. NIK may induce some essential biological functions beyond the activation of the noncanonical NF-κB pathway. NIK forms a complex with the mitochondria-associated fission protein Drp1 and phosphorylates it at serine 616 [28]. NIK also associates with the mitochondrial membrane protein PGAM5, which is required for the GTPase activity of Drp1 by dephosphorylating the serine 637 [29]. Consequently, NIK interaction with Drp1 at the outer membrane of mitochondria may induce mitochondrial fission, which can induce oxidative stress by increasing ROS production and oxygen consumption [28,30]. Mitochondrial abnormalities, in which NIK plays important causal roles, are strongly linked to the development of peripheral insulin resistance [31,32]. Analogous to its effects on peripheral glucose metabolism, it seems plausible to predict that NIK activation in the brain would directly contribute to insulin resistance and glucose intolerance which play a key role in neurodegeneration [33,34]. Interestingly, TNFR signaling can be activated independently of TNF in the presence of oxidative stress by self-dimerization of the receptor [35]. In line with this, NIK is linked to and required for the formation of the RIPK1/FADD/caspase-8 apoptotic death complex IIb [36]. Furthermore, a recent study demonstrated that NIK attenuates the cell protective function of JAK/STAT signaling, which has been shown to protect neurons [37] by phosphorylating JAK2 [38]. It has been previously reported that oxidative stress activates c-Abl, a non-receptor tyrosine kinase, in neurons [39] TNFR2-driven NIK activation and resultant oxidative stress may cause in the threonine 735 (T735) phosphorylation and cytoplasmic translocation of c-Abl [40]. NIK has also been shown to initiate the cytoplasmic activity of c-Abl by direct phosphorylating T735 [41]. Oxidative stress, protein aggregate deposition, and damaged mitochondria are hallmarks of neurodegenerative diseases [42]. Studies have shown that aberrant c-Abl activation causes neuroinflammation by promoting all these parameters [39,43]. The following are some of the major effects of activated and cytoplasmic localized c-Abl that may contribute to neurodegenerative pathologies. 1) Neurodegenerative diseases are characterized by the proliferation of activated microglia. Microglia are activated in response to PAMP, DAMP, and other environmental stimuli. Remarkably, increased TNF secretion was observed in activated microglia due to c-Abl and c-Abl-induced PKCδ activation [44]. c-Abl also activates p38 MAPK [45], which triggers plasma membrane pore formation and shedding of TNF-containing microvesicles from microglia, contributing to unconventional TNF signaling. 2) c-Abl phosphorylates OTULIN at tyrosine 56 (Y56); this abolishes the interaction between OTULIN and LUBAC [46], facilitating the interaction between LUBAC and SPATA2-bound CYLD [47]. SPATA2 is an allosteric activator of CYLD, a deubiquitinase for K63-linked ubiquitin chains. Thus, CYLD both inhibits TNFR-mediated NF-kB activation and induces cell death signaling [48]. Furthermore, Y56 phosphorylated OTULIN interacts with β-catenin, causing aberrant Wnt/β-catenin activation [46], which is involved in the development of most neurodegenerative diseases [49]. 3) c-Abl interacts with the PSMA7, a subunit of the 20S proteasome core complex, and phosphorylates at tyrosine 153, leading to the inhibition of proteasomal activity [50]. Proteasomal dysfunction, in turn, may result in protein accumulation and aggregation, as well as a loss of proteostasis, all of which contribute to neurodegenerative diseases [51]. 4) c-Abl phosphorylates hAha1 at tyrosine 223, promoting Hsp90 association and thus increasing Hsp90 ATPase activity. Thus, Hsp90 interaction with kinase clients improves, while chaperoning of non-kinase clients is compromised [52]. Because the majority of kinase clients are involved in oncogenesis, Hsp90 has been suggested as a facilitator of "oncogene addiction"[53]. In contrast, increased Hsp90 ATPase activity reduces its binding to proteins involved in neurodegenerative disorders, such as α-synuclein, promoting aggregate formation [54]. Overall, increased binding of hAha1 to Hsp90 may contribute to the accumulation of toxic proteins and neurotoxicity [55]. 5) c-Abl phosphorylates tau at tyrosine 394 [56,57]. Tyrosine-phosphorylated tau accumulates in intracellular aggregates, implying that tyrosine phosphorylation influences tau filament formation [58]. 6) c-Abl was shown as a major regulator of Parkin function by phosphorylating it on tyrosine 143. Parkin's ubiquitin ligase and cytoprotective activities are lost because of this post-translational modification, and its substrates, PARIS in particular, accumulate [59]. 7) Aside from contributing to PARIS accumulation, c-Abl phosphorylates PARIS at tyrosine 137, which is required for PARIS-induced cytotoxicity, including inhibition of the PGC-1-NRF1 pathway [60]. PARIS protein accumulation, on the other hand, can activate c-Abl tyrosine kinase, creating a pathological feed-forward loop [61]. 8) c-Abl phosphorylates α-synuclein at tyrosine 39, facilitating aggregation [62] and prion-like propagation of α-synuclein, which is important in neurodegeneration progression [63]. 9) c-Abl potentiates p53 activity by inhibiting Mdm2, an E3 ligase that is responsible for p53 degradation [64]. Thus, cytoplasmic accumulation of p53 may repress autophagy [65]. Surprisingly, α-synucleinopathy was found to activate both c-Abl and p53, inhibiting autophagy [66]. Taken together, another feed-forward regulation between c-Abl activation and α-synuclein accumulation could exacerbate the existing pathology. 10) c-Abl phosphorylates caveolin-1, a plasma membrane protein, at tyrosine 14 (Y14) [67], which regulates lipid raft-dependent macromolecular transcytosis in neurons. Therefore, c-Abl-mediated phosphorylation of caveolin-1 is important in cell-to-cell α-synuclein transmission and promotes the formation of Lewy bodies-like inclusion bodies [68]. Given that, TNFR2 has a higher TNF transcytosis capacity than TNFR1 [69]; it is conceivable that Y14 phosphorylated caveolin-1 may further enhance unconventional TNF signaling, thus causing another of the pathological feed-forward loops.
An axiomatic approach to the aforementioned neurodegeneration data.
The current prevailing view is that when stimulated, TNFR2 promotes neuronal viability by activating both canonical and noncanonical NF-κB pathways. Given the proinflammatory effects of increased NF-B activation in neurons, the question of what the teleological advantage of simultaneously stimulating both NF-κB pathways by TNFR2 may arise. Indeed, it has been demonstrated that TNFR2 in the CNS may respond differently to various conditions and ligands, resulting in diverse outcomes. In light of the above-described detailed data, the model to be presented is more likely to be rational. In the resting state, cross-talk with glial cells, astrocytes in particular, through TNFR2-mTNF and the resultant basal NF-kB activation are required for neuronal cell viability. Microglia, CNS innate immune cells, on the other hand, mediate the cytokine-induced inflammatory response primarily through the secretion of mobile vesicles in response to danger signals. MVs, containing TmTNF, secreted by microglia may interact with neural TNFR2. Endocytosis and the translocation of the tmTNF-TNFR2 complex to the perinuclear space can activate NIK. Given that neurodegenerative diseases are caused by the presence of predisposing gene alleles (risk genes) that interact with harmful environmental stimuli [70], it is clear that unregulated NIK activation will contribute to the pathology required for neurodegeneration. Thus, aberrantly activated NIK itself is detrimental to neuronal homeostasis. Furthermore, cytoplasmic activation of c-Abl by NIK or indirectly by NIK-Drp1-mediated mitochondrial fission triggers nearly all processes required for the pathogenesis of neurodegeneration. Furthermore, the resultant pathological feedforward loops create cytotoxicity through some important dysfunctions such as mitochondrial abnormalities, ubiquitinated protein accumulation due to simultaneous proteasome and autophagy inhibition, and increased pro-death activity (Figure B). Activation of NIK and c-Abl, two main interrelated kinases that can cause neurodegeneration following cytoplasmic activity of TNFR2, brings treatment options as well. The first of these is NIK inhibition. Currently, several small molecule inhibitors targeting NIK have been developed. However, given the importance of NIK activity in systemic immune regulation, NIK inhibition does not seem like a good choice due to potential systemic adverse effects. Endogenous c-Abl kinase activity is regulated by a wide range of stimuli, including growth factors, chemokines, DNA damage, oxidative stress, and adhesion receptors, as well as microbial pathogens. In response to various growth factors and antigenic stimuli, c-Abl regulates mitogenic activity in normal cells [71]. Emerging evidence suggests that proliferative and cell cycle activities promoted by activated c-Abl are known to contribute to the development of cancer and neurodegenerative diseases [72,73]. Therefore, tyrosine kinase inhibitors (TKIs) for c-Abl are employed to suppress hyperactive kinase activity in various pathologies [71,74]. The application of second and third-generation TKIs resulted in faster and more promising responses. However, their successful outcomes are frequently accompanied by a more severe toxicity profile and drug resistance [75]. Therefore, rather than inhibiting the total activity of c-Abl, the second option should be to control its cytoplasmic activity. In the cytoplasm, c-Abl undertakes the task of preparing postmitotic cells for programmed death in the face of threatening danger signals. T735 phosphorylation of c-Abl, which does not occur in a normally functioning cell, can be considered a marker of abnormal activity that impairs cellular homeostasis. In other words, it could be anticipated that inhibiting the presence of c-Abl in the cytoplasm would not be harmful to the healthy cells of the organism. Taken together, the development of a therapeutic strategy targeting T735 phosphorylation, which initiates the pathology-inducing cytoplasmic translocation of c-Abl, appears more encouraging. Finally, T735 phosphorylation of c-Abl in the cytoplasm can simply put, promote carcinogenesis in mitotically active cells and neurodegeneration in postmitotic neurons. Therefore, future T735 phosphorylation inhibitors of c-Abl would thus have the advantage of “killing two birds with one stone,” so to speak. CONFLICT OF INTEREST The author has no conflict of interest to report
Further details
Keywords
c-Abl; Drp1; NF-κB; NIK; neurodegeneration; oxidative stress; TNF; TNFR2
Abbreviations used
cIAP: cellular inhibitor of apoptosis; CNS: central nervous system; DAMP: damage‐associated molecular pattern; Drp1: dynamin-related protein 1; E2: ubiquitin-conjugating enzyme; FADD: Fas-associated protein with death domain; hAha1: human activator of Hsp90 ATPase 1; Hsp90: heat shock protein 90; IKK: inhibitor of nuclear factor kappa B kinase; IKK: Inhibitor of nuclear factor kappa-B kinase; JAK: Janus kinase; LUBAC: linear ubiquitin chain assembly complex; NEMO: NF-kappaB essential modulator; NF-κB: nuclear factor-kappa B; NIK: NF-kappaB-inducing kinase; NIK: NF-κB-inducing kinase; NRF1: nuclear Respiratory Factor 1; OTULIN: OTU DUB with linear specificity; PAMP: Pathogen‐associated molecular pattern; PARIS: parkin interacting substrate; PGAM5: Phosphoglycerate mutase 5; PGC1: peroxisome proliferator-activated receptor gamma coactivator 1; PINK1: PTEN-induced putative kinase 1; PKCδ: Protein kinase C-delta; PSMA7: Proteasome 20S Subunit Alpha 7; RIPK1: Receptor-interacting protein kinase 1; ROS: reactive oxygen species; SPATA2: Spermatogenesis Associated 2; STAT: Signal transducer and activator of transcription; TAK1: transforming growth factor-β-activated kinase 1; TNF: tumor necrosis factor; TNFR: Tumor necrosis factor receptor; TRAF: TNF receptor-associated factor; Ubc6: E2 ubiquitin-conjugating enzyme 6
Figure legend
Figure 1. Opposed neuronal functions of TNFR2 reminiscent of Dr. Jekyll and Mr. Hyde. (A) In the resting state, TNFR2, activated by paracrine mTNF, interacts with TRAF2, which is complex with TRAF3-NIK and c-IAP. TRAF2 is also associated with Parkin/PINK1 on the mitochondrial outer membrane. cIAP ubiquitinates TRAF2 by K63-linked ubiquitin chains, which stimulates the recruitment of LUBAC-OTULIN and TAK1. LUBAC adds M1-linked ubiquitin chains, resulting in the formation of mixed K63-linked and M1-linked ubiquitin chains, which is required to recruit NEMO. Subsequent phosphorylation of NEMO- associated IKKα and IKKβ by TAK1 activates the NF-κB signaling pathway, which starts the transcription of a large number of genes, required for neuronal survival. Furthermore, the formation of the NEMO-IKKα-IKKβ complex is necessary to phosphorylate and inhibit NIK. (B). Activated microglia secretes microvesicle (MV), which contain transmembrane TNF (tmTNF). The latter interacts with TNFR2, enforcing the endocytosis of the ligand-receptor complex to the perinuclear area. Cytoplasmic TNFR2-associated TRAF2-cIAP-TRAF3-NIK meets with Ubc6 and the E2 conjugating enzyme for c-IAP at the endoplasmic membrane. cIAP ubiquitination with K48-linked ubiquitin chains promotes the degradation of TRAF2 and TRAF3, leading to NIK accumulation. NIK phosphorylates IKKα for the initiation of the noncanonical NF-κB signaling pathway. In addition, NIK phosphorylates Drp1, which facilitates Drp1-PGAM5 interaction, and thus triggers mitochondrial fission. Oxidative stress caused by mitochondrial fission or NIK itself phosphorylates c-Abl at threonine 735 (T735).Activated c-Abl phosphorylates α-synuclein and tau, stimulating their aggregate formation. Parkin's functions are inhibited by c-Abl phosphorylation. The stimulatory phosphorylation of PARIS contributes to mitochondrial abnormalities. Finally, phosphorylation of OTULIN disrupts its association with LUBAC, facilitating the interaction of CYLD and LUBAC. Black arrows for stimulation; blocked red arrows for inhibition and blue arrows for activation signals.
References
[1] Medler J, Kucka K, Wajant H (2022) Tumor Necrosis Factor Receptor 2 (TNFR2): An Emerging Target in Cancer Therapy. Cancers (Basel) 14, 2603. [2] Wu C-J, Conze DB, Li X, Ying S-X, Hanover JA, Ashwell JD (2005) TNF-α induced c-IAP1/TRAF2 complex translocation to a Ubc6-containing compartment and TRAF2 ubiquitination. EMBO J 24, 1886–1898. [3] Zarnegar BJ, Wang Y, Mahoney DJ, Dempsey PW, Cheung HH, He J, Shiba T, Yang X, Yeh W, Mak TW, Korneluk RG, Cheng G (2008) Noncanonical NF-κB activation requires coordinated assembly of a regulatory complex of the adaptors cIAP1, cIAP2, TRAF2 and TRAF3 and the kinase NIK. Nat Immunol 9, 1371–1378. [4] Borghi A, Verstrepen L, Beyaert R (2016) TRAF2 multitasking in TNF receptor-induced signaling to NF-κB, MAP kinases and cell death. Biochem Pharmacol 116, 1–10. [5] Chung J-Y, Park HR, Lee S-J, Lee S-H, Kim JS, Jung Y-S, Hwang SH, Ha N-C, Seol W-G, Lee J, Park B-J (2013) Elevated TRAF2/6 expression in Parkinson’s disease is caused by the loss of Parkin E3 ligase activity. Lab Investig 93, 663–676. [6] Ma X, Rawnsley DR, Kovacs A, Islam M, Murphy JT, Zhao C, Kumari M, Foroughi L, Liu H, Qi K, Diwan A, Hyrc K, Evans S, Satoh T, French BA, Margulies KB, Javaheri A, Razani B, Mann DL, Mani K, Diwan A (2022) TRAF2, an Innate Immune Sensor, Reciprocally Regulates Mitophagy and Inflammation to Maintain Cardiac Myocyte Homeostasis. JACC Basic to Transl Sci 7, 223–243. [7] Müller-Rischart AK, Pilsl A, Beaudette P, Patra M, Hadian K, Funke M, Peis R, Deinlein A, Schweimer C, Kuhn P-H, Lichtenthaler SF, Motori E, Hrelia S, Wurst W, Trümbach D, Langer T, Krappmann D, Dittmar G, Tatzelt J, Winklhofer KF (2013) The E3 Ligase Parkin Maintains Mitochondrial Integrity by Increasing Linear Ubiquitination of NEMO. Mol Cell 49, 908–921. [8] Cogswell PC, Kashatus DF, Keifer JA, Guttridge DC, Reuther JY, Bristow C, Roy S, Nicholson DW, Baldwin AS (2003) NF-κB and IκBα Are Found in the Mitochondria. J Biol Chem 278, 2963–2968. [9] Borghi A, Haegman M, Fischer R, Carpentier I, Bertrand MJM, Libert C, Afonina IS, Beyaert R (2018) The E3 ubiquitin ligases HOIP and cIAP1 are recruited to the TNFR2 signaling complex and mediate TNFR2-induced canonical NF-κB signaling. Biochem Pharmacol 153, 292–298. [10] Wu Z, Berlemann LA, Bader V, Sehr DA, Dawin E, Covallero A, Meschede J, Angersbach L, Showkat C, Michaelis JB, Münch C, Rieger B, Namgaladze D, Herrera MG, Fiesel FC, Springer W, Mendes M, Stepien J, Barkovits K, Marcus K, Sickmann A, Dittmar G, Busch KB, Riedel D, Brini M, Tatzelt J, Cali T, Winklhofer KF (2022) LUBAC assembles a ubiquitin signaling platform at mitochondria for signal amplification and transport of NF‐κB to the nucleus. EMBO J 41,. [11] Nan J, Hu H, Sun Y, Zhu L, Wang Y, Zhong Z, Zhao J, Zhang N, Wang Y, Wang Y, Ye J, Zhang L, Hu X, Zhu W, Wang J (2017) TNFR2 Stimulation Promotes Mitochondrial Fusion via Stat3- and NF-kB–Dependent Activation of OPA1 Expression. Circ Res 121, 392–410. [12] Razani B, Zarnegar B, Ytterberg AJ, Shiba T, Dempsey PW, Ware CF, Loo JA, Cheng G (2010) Negative Feedback in Noncanonical NF-κB Signaling Modulates NIK Stability Through IKKα-Mediated Phosphorylation. Sci Signal 3,. [13] Gray CM, Remouchamps C, McCorkell KA, Solt LA, Dejardin E, Orange JS, May MJ (2014) Noncanonical NF-κB Signaling Is Limited by Classical NF-κB Activity. Sci Signal 7,. [14] Blondeau N, Widmann C, Lazdunski M, Heurteaux C (2001) Activation of the Nuclear Factor-κB Is a Key Event in Brain Tolerance. J Neurosci 21, 4668–4677. [15] Bhakar AL, Tannis L-L, Zeindler C, Russo MP, Jobin C, Park DS, MacPherson S, Barker PA (2002) Constitutive Nuclear Factor-κB Activity Is Required for Central Neuron Survival. J Neurosci 22, 8466–8475. [16] Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, Moretti M, De Smaele E, Beg AA, Tergaonkar V, Chandel NS, Franzoso G (2011) NF-κB controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol 13, 1272–1279. [17] Díaz-García CM, Mongeon R, Lahmann C, Koveal D, Zucker H, Yellen G (2017) Neuronal Stimulation Triggers Neuronal Glycolysis and Not Lactate Uptake. Cell Metab 26, 361-374.e4. [18] Soni S, O’Dea KP, Tan YY, Cho K, Abe E, Romano R, Cui J, Ma D, Sarathchandra P, Wilson MR, Takata M (2019) ATP redirects cytokine trafficking and promotes novel membrane TNF signaling via microvesicles. FASEB J 33, 6442–6455. [19] Drago F, Lombardi M, Prada I, Gabrielli M, Joshi P, Cojoc D, Franck J, Fournier I, Vizioli J, Verderio C (2017) ATP Modifies the Proteome of Extracellular Vesicles Released by Microglia and Influences Their Action on Astrocytes. Front Pharmacol 8,. [20] Raffaele S, Lombardi M, Verderio C, Fumagalli M (2020) TNF Production and Release from Microglia via Extracellular Vesicles: Impact on Brain Functions. Cells 9, 2145. [21] Soni S, O’Dea KP, Abe E, Khamdan M, Shah S V., Sarathchandra P, Wilson MR, Takata M (2022) Microvesicle-Mediated Communication Within the Alveolar Space: Mechanisms of Uptake by Epithelial Cells and Alveolar Macrophages. Front Immunol 13,. [22] Li X, Yang Y, Ashwell JD (2002) TNF-RII and c-IAP1 mediate ubiquitination and degradation of TRAF2. Nature 416, 345–347. [23] Yang X-D, Sun S-C (2015) Targeting signaling factors for degradation, an emerging mechanism for TRAF functions. Immunol Rev 266, 56–71. [24] Sun S-C (2017) The non-canonical NF-κB pathway in immunity and inflammation. Nat Rev Immunol 17, 545–558. [25] Razani B, Reichardt AD, Cheng G (2011) Non-canonical NF-κB signaling activation and regulation: principles and perspectives. Immunol Rev 244, 44–54. [26] Marienfeld R, May MJ, Berberich I, Serfling E, Ghosh S, Neumann M (2003) RelB Forms Transcriptionally Inactive Complexes with RelA/p65. J Biol Chem 278, 19852–19860. [27] Mao X, Phanavanh B, Hamdan H, Moerman-Herzog AM, Barger SW (2016) NFκB-inducing kinase inhibits NFκB activity specifically in neurons of the CNS. J Neurochem 137, 154–163. [28] Jung J-U, Ravi S, Lee DW, McFadden K, Kamradt ML, Toussaint LG, Sitcheran R (2016) NIK/MAP3K14 Regulates Mitochondrial Dynamics and Trafficking to Promote Cell Invasion. Curr Biol 26, 3288–3302. [29] Willmann KL, Sacco R, Martins R, Garncarz W, Krolo A, Knapp S, Bennett KL, Boztug K (2016) Expanding the Interactome of the Noncanonical NF-κB Signaling Pathway. J Proteome Res 15, 2900–2909. [30] Kamradt ML, Jung J-U, Pflug KM, Lee DW, Fanniel V, Sitcheran R (2021) NIK promotes metabolic adaptation of glioblastoma cells to bioenergetic stress. Cell Death Dis 12, 271. [31] Sheng L, Zhou Y, Chen Z, Ren D, Cho KW, Jiang L, Shen H, Sasaki Y, Rui L (2012) NF-κB–inducing kinase (NIK) promotes hyperglycemia and glucose intolerance in obesity by augmenting glucagon action. Nat Med 18, 943–949. [32] Liu Y, Sheng L, Xiong Y, Shen H, Liu Y, Rui L (2017) Liver NF-κB-Inducing Kinase Promotes Liver Steatosis and Glucose Counterregulation in Male Mice With Obesity. Endocrinology 158, 1207–1216. [33] Maciejczyk M, Żebrowska E, Chabowski A (2019) Insulin Resistance and Oxidative Stress in the Brain: What’s New? Int J Mol Sci 20, 874. [34] Erol A (2013) Contribution of Insulin Resistance in Pathogenesis of Alzheimer Disease. In Metabolic Syndrome and Neurological Disorders John Wiley & Sons Ltd, Chichester, UK, pp. 51–73. [35] Ozsoy HZ, Sivasubramanian N, Wieder ED, Pedersen S, Mann DL (2008) Oxidative Stress Promotes Ligand-independent and Enhanced Ligand-dependent Tumor Necrosis Factor Receptor Signaling. J Biol Chem 283, 23419–23428. [36] Boutaffala L, Bertrand MJM, Remouchamps C, Seleznik G, Reisinger F, Janas M, Bénézech C, Fernandes MT, Marchetti S, Mair F, Ganeff C, Hupalowska A, Ricci J-E, Becher B, Piette J, Knolle P, Caamano J, Vandenabeele P, Heikenwalder M, Dejardin E (2015) NIK promotes tissue destruction independently of the alternative NF-κB pathway through TNFR1/RIP1-induced apoptosis. Cell Death Differ 22, 2020–2033. [37] Nicolas CS, Amici M, Bortolotto ZA, Doherty A, Csaba Z, Fafouri A, Dournaud P, Gressens P, Collingridge GL, Peineau S (2013) The role of JAK-STAT signaling within the CNS. JAK-STAT 2, e22925. [38] Vesting AJ, Jais A, Klemm P, Steuernagel L, Wienand P, Fog-Tonnesen M, Hvid H, Schumacher A, Kukat C, Nolte H, Georgomanolis T, Altmüller J, Pasparakis M, Schmidt A, Krüger M, Supprian MS, Waisman A, Straub BK, Raschzok N, Bernier M, Birkenfeld AL, Hövelmeyer N, Brüning JC, Wunderlich FT (2022) NIK/MAP3K14 in hepatocytes orchestrates NASH to hepatocellular carcinoma progression via JAK2/STAT5 inhibition. Mol Metab 66, 101626. [39] Alvarez AR, Sandoval PC, Leal NR, Castro PU, Kosik KS (2004) Activation of the neuronal c-Abl tyrosine kinase by amyloid-β-peptide and reactive oxygen species. Neurobiol Dis 17, 326–336. [40] Nihira K, Taira N, Miki Y, Yoshida K (2008) TTK/Mps1 controls nuclear targeting of c-Abl by 14-3-3-coupled phosphorylation in response to oxidative stress. Oncogene 27, 7285–7295. [41] Mazzera L, Abeltino M, Lombardi G, Cantoni AM, Ria R, Ricca M, Saltarella I, Naponelli V, Rizzi FMA, Perris R, Corradi A, Vacca A, Bonati A, Lunghi P (2019) Functional interplay between NF-κB-inducing kinase and c-Abl kinases limits response to Aurora inhibitors in multiple myeloma. Haematologica 104, 2465–2481. [42] Yan MH, Wang X, Zhu X (2013) Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 62, 90–101. [43] Gonfloni S, Maiani E, Di Bartolomeo C, Diederich M, Cesareni G (2012) Oxidative Stress, DNA Damage, and c-Abl Signaling: At the Crossroad in Neurodegenerative Diseases? Int J Cell Biol 2012, 1–7. [44] Lawana V, Singh N, Sarkar S, Charli A, Jin H, Anantharam V, Kanthasamy AG, Kanthasamy A (2017) Involvement of c-Abl Kinase in Microglial Activation of NLRP3 Inflammasome and Impairment in Autolysosomal System. J Neuroimmune Pharmacol 12, 624–660. [45] Pandey P, Raingeaud J, Kaneki M, Weichselbaum R, Davis RJ, Kufe D, Kharbanda S (1996) Activation of p38 Mitogen-activated Protein Kinase by c-Abl-dependent and -independent Mechanisms. J Biol Chem 271, 23775–23779. [46] Wang W, Li M, Ponnusamy S, Chi Y, Xue J, Fahmy B, Fan M, Miranda-Carboni GA, Narayanan R, Wu J, Wu Z-H (2020) ABL1-dependent OTULIN phosphorylation promotes genotoxic Wnt/β-catenin activation to enhance drug resistance in breast cancers. Nat Commun 11, 3965. [47] Douglas T, Saleh M (2019) Post-translational Modification of OTULIN Regulates Ubiquitin Dynamics and Cell Death. Cell Rep 29, 3652-3663.e5. [48] Wagner SA, Satpathy S, Beli P, Choudhary C (2016) SPATA 2 links CYLD to the TNF ‐α receptor signaling complex and modulates the receptor signaling outcomes. EMBO J 35, 1868–1884. [49] Kahn M (2014) Can we safely target the WNT pathway? Nat Rev Drug Discov 13, 513–532. [50] Liu X, Huang W, Li C, Li P, Yuan J, Li X, Qiu X-B, Ma Q, Cao C (2006) Interaction between c-Abl and Arg Tyrosine Kinases and Proteasome Subunit PSMA7 Regulates Proteasome Degradation. Mol Cell 22, 317–327. [51] Thibaudeau TA, Anderson RT, Smith DM (2018) A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat Commun 9, 1097. [52] Dunn DM, Woodford MR, Truman AW, Jensen SM, Schulman J, Caza T, Remillard TC, Loiselle D, Wolfgeher D, Blagg BSJ, Franco L, Haystead TA, Daturpalli S, Mayer MP, Trepel JB, Morgan RML, Prodromou C, Kron SJ, Panaretou B, Stetler-Stevenson WG, Landas SK, Neckers L, Bratslavsky G, Bourboulia D, Mollapour M (2015) c-Abl Mediated Tyrosine Phosphorylation of Aha1 Activates Its Co-chaperone Function in Cancer Cells. Cell Rep 12, 1006–1018. [53] Neckers L, Workman P (2012) Hsp90 Molecular Chaperone Inhibitors: Are We There Yet? Clin Cancer Res 18, 64–76. [54] Daturpalli S, Waudby CA, Meehan S, Jackson SE (2013) Hsp90 Inhibits α-Synuclein Aggregation by Interacting with Soluble Oligomers. J Mol Biol 425, 4614–4628. [55] Shelton LB, Baker JD, Zheng D, Sullivan LE, Solanki PK, Webster JM, Sun Z, Sabbagh JJ, Nordhues BA, Koren J, Ghosh S, Blagg BSJ, Blair LJ, Dickey CA (2017) Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc Natl Acad Sci 114, 9707–9712. [56] Derkinderen P (2005) Tyrosine 394 Is Phosphorylated in Alzheimer’s Paired Helical Filament Tau and in Fetal Tau with c-Abl as the Candidate Tyrosine Kinase. J Neurosci 25, 6584–6593. [57] Tremblay MA, Acker CM, Davies P (2010) Tau Phosphorylated at Tyrosine 394 is Found in Alzheimer’s Disease Tangles and can be a Product of the Abl-Related Kinase, Arg. J Alzheimer’s Dis 19, 721–733. [58] Vega IE, Cui L, Propst JA, Hutton ML, Lee G, Yen S-H (2005) Increase in tau tyrosine phosphorylation correlates with the formation of tau aggregates. Mol Brain Res 138, 135–144. [59] Ko HS, Lee Y, Shin J-H, Karuppagounder SS, Gadad BS, Koleske AJ, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM (2010) Phosphorylation by the c-Abl protein tyrosine kinase inhibits parkin’s ubiquitination and protective function. Proc Natl Acad Sci 107, 16691–16696. [60] Kim H, Shin J-Y, Jo A, Kim JH, Park S, Choi J-Y, Kang HC, Dawson VL, Dawson TM, Shin J-H, Lee Y (2021) Parkin interacting substrate phosphorylation by c-Abl drives dopaminergic neurodegeneration. Brain 144, 3674–3691. [61] Giesert F (2021) c-Abl phosphorylation primes PARIS for neurodegeneration. Brain 144, 3555–3557. [62] Brahmachari S, Ge P, Lee SH, Kim D, Karuppagounder SS, Kumar M, Mao X, Shin JH, Lee Y, Pletnikova O, Troncoso JC, Dawson VL, Dawson TM, Ko HS (2016) Activation of tyrosine kinase c-Abl contributes to α-synuclein–induced neurodegeneration. J Clin Invest 126, 2970–2988. [63] Luk KC, Kehm V, Carroll J, Zhang B, O’Brien P, Trojanowski JQ, Lee VM-Y (2012) Pathological α-Synuclein Transmission Initiates Parkinson-like Neurodegeneration in Nontransgenic Mice. Science (80- ) 338, 949–953. [64] Goldberg Z (2002) Tyrosine phosphorylation of Mdm2 by c-Abl: implications for p53 regulation. EMBO J 21, 3715–3727. [65] Tasdemir E, Maiuri MC, Galluzzi L, Vitale I, Djavaheri-Mergny M, D’Amelio M, Criollo A, Morselli E, Zhu C, Harper F, Nannmark U, Samara C, Pinton P, Vicencio JM, Carnuccio R, Moll UM, Madeo F, Paterlini-Brechot P, Rizzuto R, Szabadkai G, Pierron G, Blomgren K, Tavernarakis N, Codogno P, Cecconi F, Kroemer G (2008) Regulation of autophagy by cytoplasmic p53. Nat Cell Biol 10, 676–687. [66] Karim MR, Liao EE, Kim J, Meints J, Martinez HM, Pletnikova O, Troncoso JC, Lee MK (2020) α-Synucleinopathy associated c-Abl activation causes p53-dependent autophagy impairment. Mol Neurodegener 15, 27. [67] Sanguinetti AR, Mastick CC (2003) c-Abl is required for oxidative stress-induced phosphorylation of caveolin-1 on tyrosine 14. Cell Signal 15, 289–298. [68] Ha T-Y, Choi YR, Noh HR, Cha S-H, Kim J-B, Park SM (2021) Age-related increase in caveolin-1 expression facilitates cell-to-cell transmission of α-synuclein in neurons. Mol Brain 14, 122. [69] Pan W, Tu H, Yu C, Hsuchou H, Yang Y, Kastin A (2007) Differential Role of TNF Receptors in Cellular Trafficking of Intact TNF. Cell Physiol Biochem 20, 559–568. [70] de Haan P, Klein HC, ’t Hart BA (2017) Autoimmune Aspects of Neurodegenerative and Psychiatric Diseases: A Template for Innovative Therapy. Front Psychiatry 8,. [71] Sirvent A, Benistant C, Roche S (2008) Cytoplasmic signalling by the c-Abl tyrosine kinase in normal and cancer cells. Biol Cell 100, 617–631. [72] Erol A (2010) Are Paradoxical Cell Cycle Activities in Neurons and Glia Related to the Metabolic Theory of Alzheimer’s Disease? J Alzheimer’s Dis 19, 129–135. [73] Hantschel O, Superti-Furga G (2004) Regulation of the c-Abl and Bcr–Abl tyrosine kinases. Nat Rev Mol Cell Biol 5, 33–44. [74] Khatri A, Wang J, Pendergast AM (2016) Multifunctional Abl kinases in health and disease. J Cell Sci 129, 9–16. [75] Vener C, Banzi R, Ambrogi F, Ferrero A, Saglio G, Pravettoni G, Sant M (2020) First-line imatinib vs second- and third-generation TKIs for chronic-phase CML: a systematic review and meta-analysis. Blood Adv 4, 2723–2735. [76] Erol A (2022) Genotoxicity-Stimulated and CYLD-Driven Malignant Transformation. Cancer Manag Res Volume 14, 2339–2356.
Stats
- Recommendations n/a n/a positive of 0 vote(s)
- Views 370
- Comments 0